Estimating the boiling point of water
January 01, 2012 at 07:40 PM | categories: uncategorized | View Comments
Contents
Estimating the boiling point of water
John Kitchin
I got distracted looking for Shomate parameters for ethane today, and came across this website on predicting the boiling point of water using the Shomate equations. The basic idea is to find the temperature where the Gibbs energy of water as a vapor is equal to the Gibbs energy of the liquid.
clear all; close all
Liquid water
http://webbook.nist.gov/cgi/cbook.cgi?ID=C7732185&Units=SI&Mask=2#Thermo-Condensed valid over 298-500
Hf_liq = -285.830; % kJ/mol S_liq = 0.06995; % kJ/mol/K shomateL = [-203.6060 1523.290 -3196.413 2474.455 3.855326 -256.5478 -488.7163 -285.8304];
Gas phase water
http://webbook.nist.gov/cgi/cbook.cgi?ID=C7732185&Units=SI&Mask=1&Type=JANAFG&Table=on#JANAFG
Interestingly, these parameters are listed as valid only above 500K. That means we have to extrapolate the values down to 298K. That is risky for polynomial models, as they can deviate substantially outside the region they were fitted to.
Hf_gas = -241.826; % kJ/mol S_gas = 0.188835; % kJ/mol/K shomateG = [30.09200 6.832514 6.793435 -2.534480 0.082139 -250.8810 223.3967 -241.8264];
Compute G over a range of temperatures.
T = linspace(0,200)' + 273.15; % temperature range from 0 to 200 degC t = T/1000; % normalize sTT by 1/1000 so entropies are in kJ/mol/K sTT = [log(t) t t.^2/2 t.^3/3 -1./(2*t.^2) 0*t.^0 t.^0 0*t.^0]/1000; hTT = [t t.^2/2 t.^3/3 t.^4/4 -1./t 1*t.^0 0*t.^0 -1*t.^0]; Gliq = Hf_liq + hTT*shomateL - T.*(sTT*shomateL); Ggas = Hf_gas + hTT*shomateG - T.*(sTT*shomateG);
Interpolate the difference between the two vectors to find the boiling point
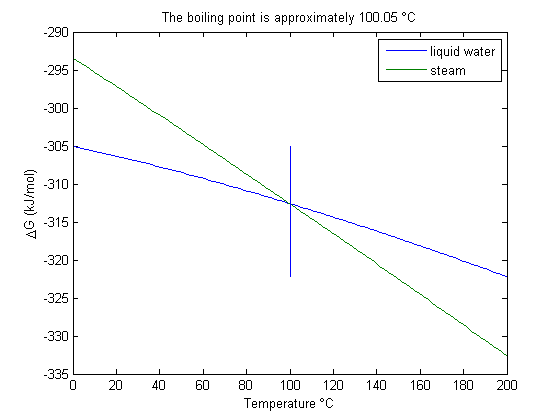
The boiling point is where Gliq = Ggas, so we solve Gliq(T)-Ggas(T)=0 to find the point where the two free energies are equal.
f = @(t) interp1(T,Gliq-Ggas,t); bp = fzero(f,373)
bp = 373.2045
Plot the two free energies
You can see the intersection occurs at approximately 100 degC.
plot(T-273.15,Gliq,T-273.15,Ggas) line([bp bp]-273.15,[min(Gliq) max(Gliq)]) legend('liquid water','steam') xlabel('Temperature \circC') ylabel('\DeltaG (kJ/mol)') title(sprintf('The boiling point is approximately %1.2f \\circC', bp-273.15))
Summary
The answer we get us 0.05 K too high, which is not bad considering we estimated it using parameters that were fitted to thermodynamic data and that had finite precision and extrapolated the steam properties below the region the parameters were stated to be valid for.
% tags: thermodynamics
 ,
,  ,
, 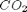 and
and 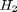 , at 1000K, and 10 atm total pressure with an initial equimolar molar flow rate of
, at 1000K, and 10 atm total pressure with an initial equimolar molar flow rate of 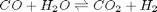 . Compared to the value of K = 1.44 we found at the end of
. Compared to the value of K = 1.44 we found at the end of 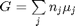 . Recalling that we define
. Recalling that we define 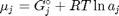 , and in the ideal gas limit,
, and in the ideal gas limit, 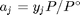 , and that
, and that 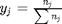 . Since in this problem, P = 1 atm, this leads to the function
. Since in this problem, P = 1 atm, this leads to the function 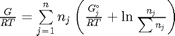 .
.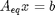 where
where  is a vector of the moles of each species present in the mixture. CH4 C2H4 C2H2 CO2 CO O2 H2 H2O C2H6
is a vector of the moles of each species present in the mixture. CH4 C2H4 C2H2 CO2 CO O2 H2 H2O C2H6 . Even though nearly all the ethane is consumed, we don't get the full yield of hydrogen. It appears that another equilibrium, one between CO, CO2, H2O and H2, may be limiting that, since the rest of the hydrogen is largely in the water. It is also of great importance that we have not said anything about reactions, i.e. how these products were formed. This is in stark contrast to
. Even though nearly all the ethane is consumed, we don't get the full yield of hydrogen. It appears that another equilibrium, one between CO, CO2, H2O and H2, may be limiting that, since the rest of the hydrogen is largely in the water. It is also of great importance that we have not said anything about reactions, i.e. how these products were formed. This is in stark contrast to 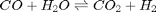 . We can compute the Gibbs free energy of the reaction from the heats of formation of each species. Assuming these are the formation energies at 1000K, this is the reaction free energy at 1000K.
. We can compute the Gibbs free energy of the reaction from the heats of formation of each species. Assuming these are the formation energies at 1000K, this is the reaction free energy at 1000K.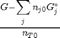
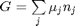 where
where 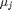 is the chemical potential of species
is the chemical potential of species  , and it is temperature and pressure dependent, and
, and it is temperature and pressure dependent, and 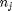 is the number of moles of species
is the number of moles of species 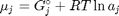 , where
, where 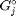 is the Gibbs energy in a standard state, and
is the Gibbs energy in a standard state, and  is the activity of species
is the activity of species  and stoichiometric coefficients:
and stoichiometric coefficients: 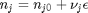 . Note that the reaction extent has units of moles.
. Note that the reaction extent has units of moles.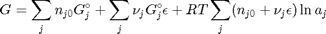
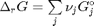 . With these observations we rewrite the equation as:
. With these observations we rewrite the equation as: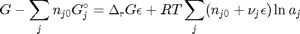
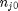 . It is desirable to avoid this, so we now rescale the equation by the total initial moles present,
. It is desirable to avoid this, so we now rescale the equation by the total initial moles present, 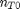 and define a new variable
and define a new variable 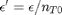 , which is dimensionless. This leads to:
, which is dimensionless. This leads to: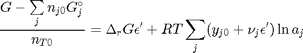
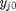 is the initial mole fraction of species
is the initial mole fraction of species 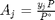 , where the numerator is the partial pressure of species
, where the numerator is the partial pressure of species 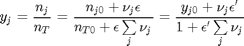
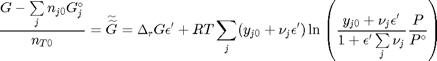
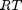 . This additional scaling makes the quantities dimensionless, and makes the quantity have a magnitude of order unity, but otherwise has no effect on the shape of the graph.
. This additional scaling makes the quantities dimensionless, and makes the quantity have a magnitude of order unity, but otherwise has no effect on the shape of the graph.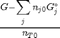
 can be. Recall that
can be. Recall that 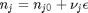 , and that
, and that 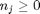 . Thus,
. Thus, 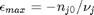 , and the maximum value that
, and the maximum value that 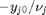 where
where 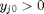 . When there are multiple species, you need the smallest
. When there are multiple species, you need the smallest 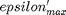 to avoid getting negative mole numbers.
to avoid getting negative mole numbers.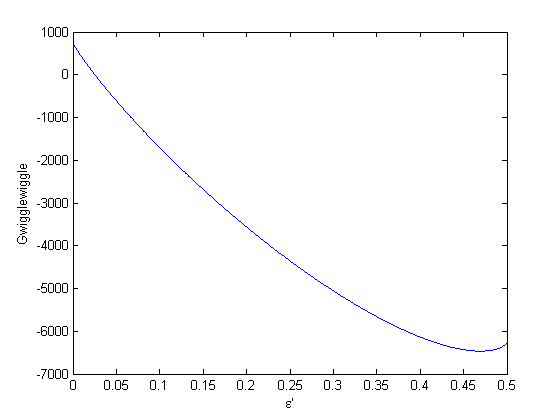
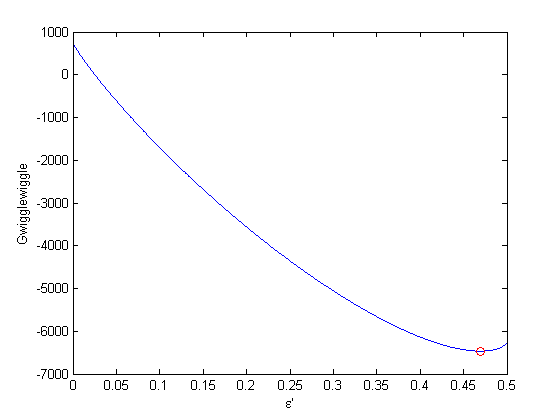
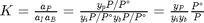 .
.